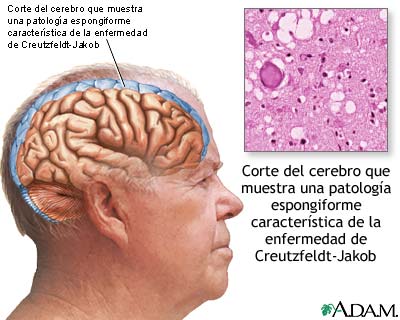
LA enfermedad de Creutzfeldt-Jakob (CJD) es la enfermedad priónica humana más común, caracterizada por demencia rápidamente progresiva, trastorno de la marcha, postura rígida, ataques epilépticos y parálisis facial que le da al individuo afectado la apariencia de estar siempre sonriendo. Esta enfermedad pertenece al grupo de encefalopatías espongiformes.
La ECJ se describió por primera vez en 1920 y en 1968 se transmitió con éxito a un chimpancé, lo que demuestra la existencia de un agente infeccioso. Sin embargo, este agente era resistente a las técnicas que inactivaban los ácidos nucleicos, pero no a las técnicas que resultaban en proteólisis.
En la década de 1980 se detectó la presencia de una determinada proteína en los tejidos de pacientes con encefalopatías espongiformes, denominada PrP (proteína resistente a proteinasa). Prussiner planteó la hipótesis de que este era el agente infeccioso e instituyó el término prión. Investigaciones realizadas posteriormente demostraron que la proteína PrP, junto con los procesos patológicos (PrPres), era una isoforma de una proteína generalmente sintetizada por el propio huésped (PrPsen), ubicada en el cromosoma 20. En condiciones relacionadas con los priones, PrPsen sufre una alteración estructura, siendo la nueva forma se acumula en las neuronas, dando lugar a la muerte celular.
Actualmente existen cuatro formas de esta enfermedad, que son:
- Esporádica: esta forma representa el 85% de los casos, con una incidencia de 0,5 a 1,5 casos por millón cada año. No hay variación estacional en los casos ni factores de riesgo relacionados.
- Familiar: representa el 10-15% de los casos y tiene un patrón de transmisión autosómico dominante.
- Iatrogénica: esta forma se observó en 1974 después de un trasplante de córnea y se ha descrito desde entonces con el uso de injertos de duramadre, la administración de hormonas de crecimiento humano y el uso de instrumentos en neurocirugías inadecuadamente esterilizadas.
- Nueva variante: esta forma se llama encefalopatía espongiforme (EEB, más conocida como “enfermedad de las vacas locas”), que se mencionó anteriormente. Hay pocos casos descritos de esta manera.
Ocurre con mayor frecuencia en personas de entre 50 y 70 años (80% de los casos). El inicio de las manifestaciones clínicas varía, y un tercio de los individuos se caracteriza por síntomas vagos como fatiga, trastornos del sueño o anorexia. Otro tercio comienza con amnesia, confusión mental o cambios de comportamiento. El grupo restante presenta signos focales como afasia, hemiparesia, ataxia o amiotrofia. La progresión de esta enfermedad es rápida, provocando degeneración cognitiva y aparición de mioclonías, pudiendo presentarse también otros signos clínicos como coreoatetosis, ataxia o síntomas de afectación de la segunda motoneurona. Al final de la afección, el paciente se vuelve acinético y es posible que ya no presente mioclonías. El tiempo medio de supervivencia es de alrededor de 5 meses y aproximadamente el 80% de los pacientes evolucionan hacia la muerte en menos de un año.
Normalmente, al inicio de la afección, el electroencefalograma es normal, pero a medida que avanza la afección, en más del 80% de los pacientes este examen revela descargas agudas periódicas, generalmente trifásicas y sincrónicas de 0,5 Hz en algún momento del período. el desorden.
Al igual que con el electroencefalograma, las pruebas de imagen como la tomografía computarizada y la resonancia magnética pueden presentar resultados normales al inicio de la enfermedad y, a medida que avanza, muestran atrofia cerebral generalizada e hiperdensidad en los ganglios linfáticos de la base.
En cuanto al diagnóstico de laboratorio, este no muestra cambios en las pruebas de actividad inflamatoria o anticuerpos que neutralizan al agente. En raras ocasiones, se puede observar un ligero aumento de proteínas en el líquido cefalorraquídeo. Esta proteína tiene una sensibilidad del 96% y una especificidad del 99% para la ECJ.
La mejor forma de diagnosticar esta enfermedad es mediante el análisis histopatológico del tejido cerebral con tinción inmunohistoquímica para PrPres. Los hallazgos más importantes son:
- Alteraciones espongiformes;
- Astrocitosis;
- pérdida neural
- Las placas mieloides se observan en el 10% de los casos de forma esporádica.
No existe cura para la enfermedad, ni una terapia capaz de retrasar la evolución de la enfermedad. La única opción es realizar medidas paliativas mediante tratamiento sintomático. Como esta enfermedad es contagiosa, es necesario evitar el trasplante o la ingestión de tejido infectado.
Fuentes:
http://pt.wikipedia.org/wiki/Doen%C3%A7a_de_Creutzfeldt-Jakob
http://www.manualmerck.net/?id=212&cn=1798
http://adam.sertaoggi.com.br/encyclopedia/ency/article/000788.htm
http://www.unifesp.br/dneuro/neurociencias/217_relato.pdf
http://www.medcenter.com/medscape/Content.aspx?id=545
http://virtualpsy.locaweb.com.br/index.php?art=79&sec=16