
LA Enfermedad de Gaucher es una condición de origen genético relacionada con la deposición lisosomal, caracterizada por la acumulación de glucosilceramida en macrófagos / monocitos. Esta enfermedad forma parte de un grupo en el que participan más de 40 enfermedades genéticas, caracterizadas por la ausencia de una o más enzimas, afectando a 1 de cada 50.000 a 100.000 individuos.
Ocurre debido a una deficiencia de la enzima lisosomal ácida β-glucosidasa, también conocida como glucocerebrosidasa. Su tarea, en individuos libres de enfermedades, es descomponer un sustrato lipídico, el glucocerebrósido, dentro de la célula. Como consecuencia de la alteración en el gen responsable de producir la enzima en cuestión, su cantidad es insuficiente y no tiene la capacidad de descomponer el sustrato a la velocidad ideal, comenzando a acumularse en los ribosomas.
Los lisosomas ubicados dentro de los macrófagos son orgánulos responsables de eliminar los restos celulares. Sin embargo, debido a la acumulación de sustrato lipídico en estas estructuras, los macrófagos son incapaces de llevar a cabo su función, comenzando a recibir el nombre de células de Gaucher.
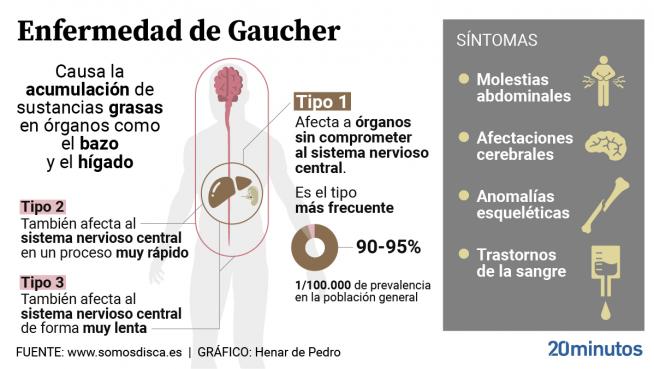
Estas células tienden a acumularse en el hígado, el bazo, los pulmones y la médula ósea; Los riñones, los ganglios linfáticos y la piel también pueden verse afectados. Con menos frecuencia, la acumulación se encuentra en los tejidos del sistema nervioso central. Los órganos afectados suelen hipertrofiar, dando lugar a manifestaciones clínicas variables.
Se describen tres tipos de enfermedad:
- Tipo 1 o forma no neuropática: este es el tipo más común y representa aproximadamente el 90% de los casos. Afecta a niños y adultos, con síntomas de aparición variable. Las manifestaciones clínicas / signos clínicos que suelen presentarse son hepatomegalia, esplenomegalia, avanzando a hiperesplenismo con anemia progresiva, trombocitopenia y leucopenia. Esta condición puede ir acompañada de fatiga, cansancio, plenitud gástrica posprandial y retraso del crecimiento en los niños. La acumulación de sustrato lipídico en la médula ósea provoca osteopenia, lesiones líticas, fracturas patológicas, dolor óseo crónico, crisis óseas, infarto y osteonecrosis. También se observó una mayor incidencia de neoplasias óseas en pacientes con esta enfermedad. La progresión de la afección varía y la supervivencia puede estar dentro del rango normal.
- Tipo 2 o forma neuropática aguda: en este tipo existe un deterioro severo del sistema nervioso central y síntomas tempranos en la infancia. Afecta a lactantes de 4 a 5 meses de vida, afectando el cerebro, el hígado y los pulmones. El cuadro neurológico es severo, con convulsiones repetidas, hipertonía, apnea y retraso mental progresivo. La evolución se acelera y progresa hasta la muerte dentro de los dos primeros años de vida, generalmente debido a un deterioro pulmonar.
- Tipo 3 o forma neuropática crónica: afecta a niños y adolescentes, generalmente a partir de la edad preescolar. Afecta al cerebro, el bazo, el hígado y los huesos. La evolución del cuadro neurológico varía, pero es más leve que la del tipo 2. La supervivencia ronda los 20 a 30 años.
El diagnóstico más confiable se realiza midiendo la actividad de la enzima β-glucosidasa en leucocitos o fibroblastos. Cuando hay dudas sobre el diagnóstico, se puede realizar una biopsia de médula ósea o un mielograma, donde se pueden encontrar células de Gaucher.
Se desarrolló un tratamiento basado en la extracción de enzimas presentes en la placenta humana, que comenzó a utilizarse en 1991. Recién en 1994 se comenzó a producir la enzima de forma sintética, mediante la técnica del ADN recombinante, permitiendo su mayor diseminación.
Hoy en día se utilizan nuevos enfoques terapéuticos, denominados terapia de reducción de sustrato, que se utilizan por vía oral. Esta terapia actúa reduciendo la síntesis de lípidos, evitando su acumulación.
Fuentes:
http://pt.wikipedia.org/wiki/ Enfermedad de Gaucher
http://www.doencadegaucher.com.br/pacientes/conheca_gaucher/que_e_gaucher.aspx
http://portal.saude.gov.br/portal/arquivos/pdf/gaucher_pcdt.pdf
http://apps.einstein.br/revista/arquivos/PDF/527-Einstein5-1_Online_IM527_pg78-79.pdf


