
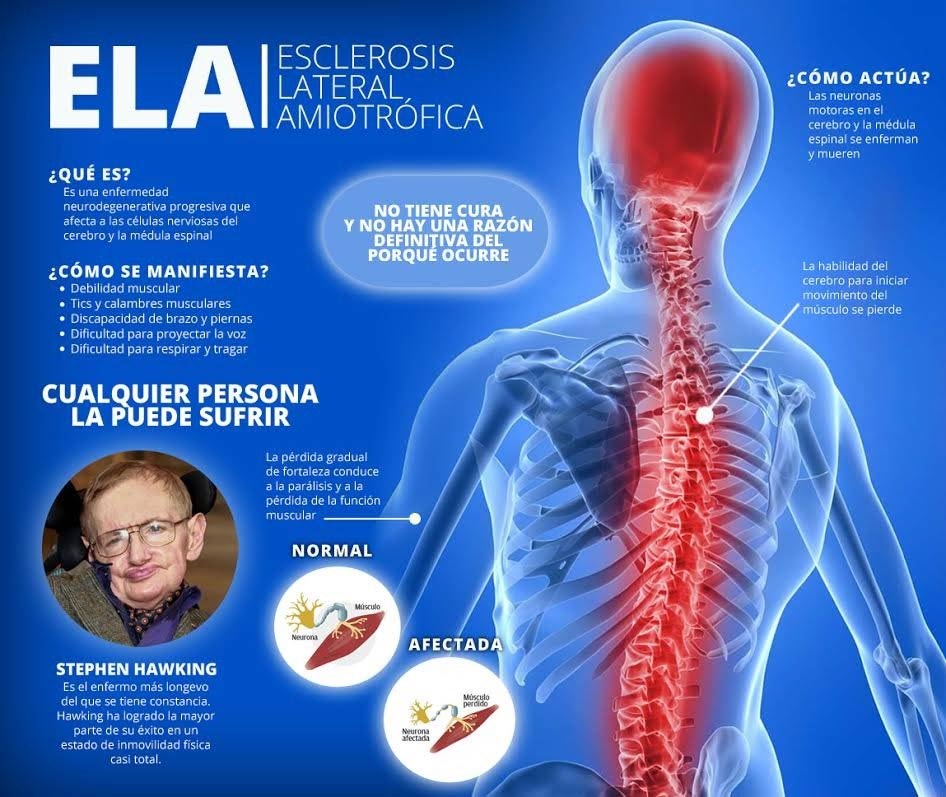
LA La esclerosis lateral amiotrófica (ELA) también conocida como enfermedad de Charcot (Francia) o enfermedad de Lou Gehring (EE. UU.) Es una enfermedad neurodegenerativa adquirida de causa desconocida, que afecta principalmente a las neuronas motoras de la médula espinal, el tronco encefálico y el cerebro. Esta enfermedad afecta al sistema nervioso de forma degenerativa y progresiva provocando una parálisis motora irreversible. La parálisis es gradual y la muerte del afectado es el resultado de la pérdida de capacidades vitales y cruciales, como hablar, moverse, tragar e incluso respirar. La incidencia de ELA – promedio de aproximadamente 1 / 50.000 por año y prevalencia – promedio de 1 / 20.000, es relativamente uniforme en los países occidentales, a pesar de haber descrito brotes más frecuentes en el Pacífico occidental. En general, hay una ligera preponderancia masculina (proporción de hombres a mujeres de aproximadamente 1,5: 1). En 2014, el Ministerio de Salud amplió la atención a las personas con enfermedades raras, instituyendo la Política Nacional de Atención Integral a las Personas con Enfermedades Raras, incluida la ELA. El Protocolo clínico y las guías terapéuticas para esta enfermedad se actualizaron en noviembre de 2015.
La ELA se puede clasificar según los primeros signos clínicos que se presenten; en cuanto a la edad de aparición de los síntomas; con respecto a la variabilidad genética; en cuanto al tiempo de progresión; y el más habitual, por el modo de herencia (familiar, esporádico y Guam). Se ha descrito que numerosos genes están asociados con la ELA. La ELA familiar representa entre el 5% y el 10% de los casos y se puede clasificar según el modo de herencia: autosómica dominante, autosómica recesiva y ligada a la X dominante. Pertenecer a este tipo de ELA significa que el paciente tiene al menos una persona afectada en la familia. La gran mayoría de los casos están relacionados con las formas dominantes y con la aparición en adultos.
Aunque existen algunas variaciones en las manifestaciones clínicas, en el patrón de progresión de la enfermedad y en la esperanza de vida después del inicio de los síntomas, el diagnóstico se realiza mediante examen clínico. El diagnóstico de ELA es evidente en pacientes con una larga evolución de la enfermedad. El diagnóstico precoz de la enfermedad, cuando el paciente solo presenta síntomas focales en una o dos regiones (bulbar, miembro superior, tronco o miembro inferior), puede resultar difícil y dependerá de la presencia de signos en otras regiones afectadas. El tiempo promedio desde el inicio de los síntomas hasta la confirmación del diagnóstico es de aproximadamente 10 a 13 meses. El diagnóstico de ELA se basa en la presencia de signos de deterioro de las neuronas motoras inferiores y neuronas motoras superiores concomitantes en diferentes regiones. Los principales signos y síntomas de la ELA se pueden agrupar en dos grupos:
- I) signos y síntomas derivados directamente de la degeneración motoneuronal: debilidad y atrofia, fasciculaciones y calambres musculares, espasticidad, disartria, disfagia, disnea y labilidad emocional;
- ii) signos y síntomas indirectos derivados de síntomas primarios: trastornos psicológicos, trastornos del sueño, estreñimiento, sialorrea, espesamiento de las secreciones mucosas, síntomas de hipoventilación crónica y dolor.
La identificación de factores de riesgo y enfermedad en su estadio inicial y la derivación oportuna y adecuada a atención especializada confieren a la Atención Primaria un carácter esencial para un mejor resultado terapéutico y pronóstico de los casos. Para el tratamiento de la ELA, hasta ahora, solo hay un medicamento aprobado por la FDA, el riluzol, y otros medicamentos a menudo se asocian con la terapia para aliviar los síntomas y mejorar la calidad de vida del paciente. En busca de un tratamiento eficaz para la ELA, varios investigadores están utilizando células madre debido al potencial de estas células para diferenciarse en diferentes tipos de células nerviosas. en vivo y in vitro. Se ha demostrado que el trasplante de células madre es eficaz en varios modelos animales, pero aún no se han aclarado las vías biológicas subyacentes a los procesos de reparación. El objetivo clínico en la ELA también ha buscado dilucidar la interacción entre las neuronas motoras y las células no neuronales (principalmente astroglia o microglia).
Bibliografía:
1. Jerez DR. Rehabilitación en la esclerosis lateral amiotrófica: revisión de la literatura. ACTA FISIATR 2008; 15 (3): 182 – 188.
2. Ministerio de Salud – acceso: 01/02/2020 – http://www.saude.gov.br/saude-de-az/ela-esclerose-lateral-amiotrofica
3. Pardina JSM. Esclerosis lateral amiotrófica y otras enfermedades de las neuronas motoras. En: Gómes JP, (editor). Tratado de neurología clínica. Barcelona: Artes Médicas; 2008. p. 797-826.
4. Silani V, Calzarossa C, Cova L, et al. Células madre en la esclerosis lateral amiotrófica: ¿protección o reemplazo de neuronas motoras? Objetivos de medicamentos para el trastorno neurológico del SNC 2010; 9 (3): 314-2


